The headline is potentially confusing, it doesn't mean that this is the first time individual atoms were measured, but the first time this particular method has been pushed to such a high resolution that individual atoms could be distinguished.
Cryo EM is a very hot method right now to determine the three-dimensional structure of large molecules like e.g. proteins or protein complexes. Something like 10-15 years ago the best you could get was maybe 1.5 - 2 nm (these are rough numbers from memory), the results described in the article are 0.17 nm.
Further, atomic resolution is routine in electron microscopy, it's specifically Cryo EM (a sub-technique aimed at imaging proteins, which are extremely fragile compared to, say, chunks of metal or computer chips) where the advance has happened.
This is phenomenal! The title still feels a bit sensationalized, but don't they all?
Cryo EM is really cool (not just in temperature! ;-) ) technology! :) Recently I had a chance to go on a tour in the two-part CEITEC research center in Brno, a city with long history of electron microscopy manufacturing and research.
One part of the CEITEC research center does "dry" tech - chemistry, robotics, motor and machine control and chips. Of course, they have quite a lot of cutting edge electron microscopes to image various nano structures. All looking super high tech as you watch their shiny metal over the clean room window. All the machines stand on concrete slabs embedded into the hill below the building to avoid vibrations and have ultra high vacuum inside.
The other part of CEITEC does "wet" research - proteins, cells, bacteria, microorganisms. Of course also they have cutting edge, cry electron microscopes. These looked quite different to the other ones - it was a huge black cube, reaching almost the high ceiling of the room. You could not see all the glistening internal components like in the previous case. In this case they even let us to the room itself - apparently given all the volatiles in the samples, the cleanliness requirements in the room containing the machine are not as strict & it likely (my guess) needs to keep quite a bit part of the machine at very low temperatures, explaining all the covers. IIRC the software controlling the camera runs on Linux & engineers from Brno working for one of the big EM manufacturers were involved in building this enormous cryo EM machine, which was a pretty nice touch. :)
When I started grad school in 2003 there were a handful of structures approaching 0.4 nm, which is the point where you might be able to build most of the sequence into the map de novo. But those were exceptional cases.
my favorite report from grad school was the paper that had to be retracted because they "built the sequence into the map backwards" (IE, the N and C termini were switched).
When I was doing this in grad school in 2009, we were pulling down to 10 angstroms in our best 3d reconstructions, sampling something around 30k virus particles (which I and some undergrads got to pick from the chromatographs).
I'm a structural biologist that uses CryoEM to study archeal viruses. I'd be happy to answer any questions about CryoEM.
I did want to point out though that this title is misleading. The researchers didn't get an image of an atom. Instead, they reconstructed hundreds of thousands of images of the protein to determine where the atoms are almost exactly. So there's no actual image that shows an atom, as cool as that would be.
You could say the same about any image produced by a scientific instrument these days. Any astronomical image has a huge amount of data behind it, but we don't sit around saying "gee, there are no actual images, isn't that a shame".
If you don't want to call something an image, you can call it a diagram produced by a data processing program, but whatever you call it, it's disappointing if it's not provided.
Ok, since at least two of you who know what you're talking about have objected to the title, I've replaced it with the more general subtitle above. Thanks!
(Submitted title was "Cryo-electron microscopy technique sees individual atoms for first time".)
For a long time, it was the cameras. We couldn't get smaller things into focus and still preserve high-resolution images. Now that the camera problem has been solved with CMOS detectors, the next problem is likely going to be the contrast-transfer function [1]. If we can solve that (such as with an energy filter, as I suspect the authors of this paper used), we can get smaller and smaller samples in focus without losing high resolution information from the electron scattering.
"Vitrification" is a way to freeze water so that it forms a glassy solid instead of ice crystals, locking the proteins in place and so that the "ice" reflects electrons randomly like water does, and so the only large structure redirecting electrons is the protein particles.
Protein "structure" is a slippery concept. In situ measurements, molecular dynamics simulations and basic common sense pretty conclusively show that most proteins actually have a number of conformations they access during normal biological processes. In many cases, these conformations are required for the proteins normal functions. I used to work in the de novo protein field and it was a constant source of irritation to me that what we mainly designed were crystal structures, not dynamic functional proteins.
There's lots of structural information such as conserved folds that can be gleaned from Cryo and X-ray structures. Protein dynamics is an important part but structures published with these mechanisms have been proven biologically relevant by site directed mutagenesis an almost infinite number of times now.
Thank you. Would it be fair to say that crystallized protein is always in one specific confirmation? Compared to that, would vitrification have the chance (however random) to capture protein molecules in different conformations?
Yes, a crystallized protein will always be in a single conformation, or you won't be able to see it because the electron density map will be an average of all possible conformations, and therefore meaningless.
Single particle gives you the opportunity to see different conformations, but only if the data is discrete. If there's a continuous amount of conformations (think a molecular motor that's rotating) you would need nearly infinite data to resolve a nearly infinite number of conformations. If the data is less than continuous, you can image enough particles to see all the different conformations by constructing multiple models in parallel and using 3D angular searching to bin them by what conformation they are in. This is a computationally exhausting process, however.
I hate to be that guy, but your first statement is technically incorrect: you can have discrete alternate conformations superimposed in the electron density (usually 2-3 is the most that can be resolved), and you can have different conformations of multiple copies of the molecule. (I've personally worked with both, although the differences in the second case were small.) That's not even counting ensemble-based approaches to modeling the crystal structure, although that's arguably just a different way of representing the uncertainty.
All that aside, crystallization certainly biases it towards specific conformations, which single-particle EM does not.
Didn't realize that. My experience with xray crystallography is limited. Obviously there's some variance, but I always assumed it would simply be unresolvable/disordered in that case.
I'm not aware of any, but I don't think that until we're able to see proteins moving we'll actually be able to answer those questions visually. We've got a long way to go before that happens.
The samples themselves are plunge frozen into vitreous ice using liquid ethane, so they won't move much on their own. The electron beam, as well as the stage they sit on inside the microscope do cause movement of the grid/sample, however, and so on modern microscopes, what we actually record are movies so that we can correct for things like beam induced motion with software.
Exposure is a separate variable because it also causes sample degradation. Ideally, a microscope is only shooting one electron at a time at the sample (that never happens obviously), but the fewer electrons, the better. On overexposed micrographs, you can see the burn marks on the sample.
I did my undergraduate degree in Nutritional Science, realized I liked enzymes/protein structure a lot, and applied to a biochemistry department PhD program. I'm in my 3rd year and currently on/ahead of track so not having a biophysics background hasn't held me back.
I'd recommend, if that's what you wanted to do, to start with biophysics or molecular biology as those are the hardest aspects. A good computer science background is helpful as well, because a huge part is processing terabytes upon terabytes of micrographs into a finished, ab initio protein model. There's always room for new techniques there.
Personally, if I were to do it over, I'd major in the closest thing to biophysics/structural biology I could, and spend as much time in a lab doing undergraduate molecular biology research as I could.
While this is a breakthrough within the field of cryo-electron microscopy it is important to appreciate that for many questions in structural biology we also need to understand how protein structure changes over time.
With the presented method the structure sampling time seems to be O(10 s) which easily is about 10 orders of magnitudes slower than the dynamics we're interested in seeing.
A direct consequence of this is that for achieving atomic spatial resolution they needed to use a "rock-solid" protein -- one that has an exceptionally stiff structure (one that does not wiggle a lot). The presented method is great and cool, but this is a pretty severe limitation. Most proteins wiggle a lot :-).
Background: protein structure-function relationship can often be well understood only when considering the structural dynamics of the protein (key words: conformational changes, the entropic contribution to free energy).
That is, in the ideal case we would be able to measure molecular structure not only at high spatial resolution, but also at high temporal resolution.
How can one make such a measurement much faster, by ~10 orders of magnitude? By irradiating a lot of light. Via X-ray free electron lasers (XFEL).
XFEL-based techniques are expected to revolutionize structural biology (as always, also still a long way to go):
> XFEL protein crystallography not only determines high resolution structures of proteins, but also reveals the time-stamped conformational changes of proteins.
You're glossing over the advantages specific to cryo-EM though, which is it can give you a good picture of large ensembles of small things at this accuracy, with a lot less computational/interpretation hardship than X-ray crystallography or NMR. So for seeing structures of large protein complexes and how the super structure varies with heteromeric variation this is a really big deal. If you wanted to see what viral capsids in-situ looked like down to the atoms this is what you'd use. There are a lot of places this is going to be useful, there is no one method to rule them all in structural proteomics simply because nothing can offer all of the spatial and temporal scales one might need. Plus cryo would only give you the surface hull of these complexes, you'd still need a different method to fill in all the exact structures inside. Writing up these methods as if there is some competition for "best" is an entirely false impression to a lay person reading. The best is all methods improve and researchers collaborate to provide every possible scoped view. Even mass spectrometry is doing crazy things for investigating the structure of disordered proteins. You'd never use that for something you would use cryo-EM for.
Thanks for this reply and the additional level of detail. I didn't mean to imply that one method is generally better than another. The combination of a variety of methods applied to the same problem space is certainly the way to go whenever we really want to unravel molecular mechanisms.
To add on, another promising technique is ultrafast electron diffraction (UED). You can get atomic space and time resolution (sub-angstrom and 100 fs) already. It's just a matter of scaling up beam quality to the point we can study complex macromolecules. UED will also fit in a single room while the worlds only XFEL is miles long.
(I work on instrumentation improvements for XFELs and UED.)
But don't forget that these methods use class-averaging and each class represents members of a frozen structure. One of the beauties of cryoEM is that if your structure is dynamic, cryoEM will actually capture the ensemble in each class, which if you have enough images of each class you can solve each states' structure. One caveat is that if there are no local minima, you may have a huge number of states which means you won't get high resolution structures of each, but typically you'll have a few conformations that are reasonably stable. The other cool thing is you can throw in literal cell lysate and let the computers "purify" the sample for you. This allows the native environment to participate in the thermodynamic landscape of dynamic proteins, possibly showing more meaningful dynamic states.
I had the privilege to ocasionally work with Holger Stark on similar structure determination challanges and can confirm that they really pushed the limits here thanks to very smart statistical methods. The resolution heavily depends on correctly sorting/classifying the large amount (> 10k for sure) images of these small particles.
There exists a race in the structural biology community about the next big method that allows to determine structures of proteins that were hard to crystalize and it seems that CryoEM is becoming the winner in this race.
As an alternative approach, people are building large X-ray lasers that have extremly high intensity and short pulse lengths which they plan to shoot at single particles and resolve individual scattering images. This method can very likely also achieve atomic resolution of non-crystaline particles, even without the need to freeze them down.
It will be exciting times for the whole bio-chemical physics community. Congrats to Holger and the team for another great publication in Nature.
Having worked in X-ray crystallography on synchrotrons and XFELs, I can't take the single-particle XFEL concept seriously any more. Ten years ago it seemed impressive, but the EM technology has advanced so much faster - and it's a fraction of the cost. There are some neat applications of nanocrystallography, but that too seems like a niche application now, not a magic bullet for accelerating protein structure determination.
The real breakthrough here is that with this technique you don't have to crystallize the proteins anymore, which is the biggest bottleneck in X-Ray Crystallography.
Relevant; they used Cyro-EM to elucidate the structure of the SARS-CoV-2 Spike protein [1], which is a big step towards the development of a vaccine. This is a resolution of 3.5 ångström, but still a very nice feat.
3.5Å cryo-EM structures have existed for some time. The important thing coming down under 2Å or so, is that it drastically improves the quality of the molecular dynamics simulations you build of these structures.
I'm skeptical of molecular dynamics and docking studies... but the ability to clearly resolve bound drug molecules, or host-pathogen interactions, is very valuable indeed.
So, pardon my ignorance, but can we finally say that atoms are particles with concrete dimensions? Last time I checked the whole field seemed measuring everything into probabilistic and statistical terms.
We can meaningfully talk about concrete dimensions just like before. Atomic radius is mean distance from the nucleus to the boundary of the surrounding shells of electrons.
Because the outer shell of electrons is fuzzy, there are multiple ways to define radius: covalent radius, Van der Waals radius and ionic radius. In molecules atoms overlap in different ways because they share electrons and electrons interact.
There still exists fundamental uncertainty about the location of atoms and molecules. Electrons, atoms and molecules have de Broglie wavelength. For molecules with tens of atoms the wavelength is in the order of picometers.
Our understanding on the nature of atoms is not something someone suddenly "discovered". Rather, we've gathered evidence of various phenomena over the century. There are lots of models to explain the findings.
This is a bit of a long winded list of discoveries. There is no simple sophism you can utilize and say "atoms are this or that" unless you actually understand something of the background.
This Khan academy lecture is as good resource as any:
I hope somebody weighs in because ever since I read that matter is just quantum field excitations I've lost all intuition about what reality is. Everything seems to be nothing.
For better or worse, Cryo-EM doesn't settle this. The results are inherently statistical no matter what, and probably can't distinguish between one microscopic model of the atom and another. I haven't seen the equations, but both the amount of data, and the amount of processing, are profound. I work in a peripherally related area, and just one anecdotal note is that the people doing this work think nothing of a single measurement filling up a terabyte hard drive with data.
There are naturally a lot of views about this topic, but one that resonated with me (perhaps recently discussed in HN) is that science can't necessarily make things intuitive, because it has this nasty habit of telling us when our intuition is wrong.
Confirmed. I've worked with structures at close to sub-atomic resolution (0.8Å), where you can even see the "deformation" of hydrogen bonds and lone pairs in the electron density. But the actual information we have is simply a grid of electron density values averaged over an entire crystal.
Could someone confirm what we’re looking at in that picture please? In that spider web of blue points joined by blue line segments: Those tiny blue points are atoms? The blue line segments are covalent bonds? And what’s the difference between the blue and purple areas?
If those line segments are probability distributions of shared electrons, that means our high school mental model of a covalent bond (drawing a line between two atoms) is not too bad.
The line segments define surfaces around the regions of highest electron density. This is how X-ray crystallographers traditionally viewed electron density, probably because that simple mesh was the best that early computer graphics systems could handle. In this particular image they've shown nested surfaces - my guess would be at 1 (purple), 2, and 3 standard deviations above the mean, or something like that. Again, not atoms, just electron clouds, but obviously the regions around the atoms have the most electrons.
So it’s not like what I thought at all. If I understand you correctly, then each one of those bright blue balls is a cloud of electrons at the center of which is a nucleus (but we don’t see the nucleus itself). The tiny blue lines and vertices are just computer generated imagery to define the “surface” of an atom. Those hexagons with six blue balls joined by purple mesh are benzene rings? So each purple area defines a cloud of shared electrons in a benzene ring?
> computer generated imagery to define the “surface” of an atom
The grids are isosurfaces of electron density. Analogous to isolines on a map illustrating mountain terrain height.
Atoms are sticky little fuzzy balls. The fuzz, electron density, falls off exponentially. They don't have an "edge" in a non-fuzzy-object sense. Analogous to a stereotype volcano having an arbitrary perimeter. Where would I place it? Where you need climbing gear? Where biking up gets hard? In the foothills? Somewhere on the sloped plain? You place the perimeter wherever is convenient for what you're trying to use it for. People are usually interested in the densities between sticking atoms, which are several orders of magnitude down from peak density. Analogous to drawing an isoline at "the towering volcano is here high enough to start stubbing your toe on it". The picture draws isosurfaces of electron density at something like "around here one atom would 'bump' another, and you can see bonds" and at "I wrap a single atom only, but still have an interesting shape".
To show the various features of interest, one draws multiple isosurfaces. As with a terrain map. But an exponential electron density scale is harder to represent than linear height scale. And it's hard to clearly draw more than a couple of nested 3D isosurfaces. So instead of placing many iso's at linear steps, here there are just a couple, more like an order of magnitude apart.
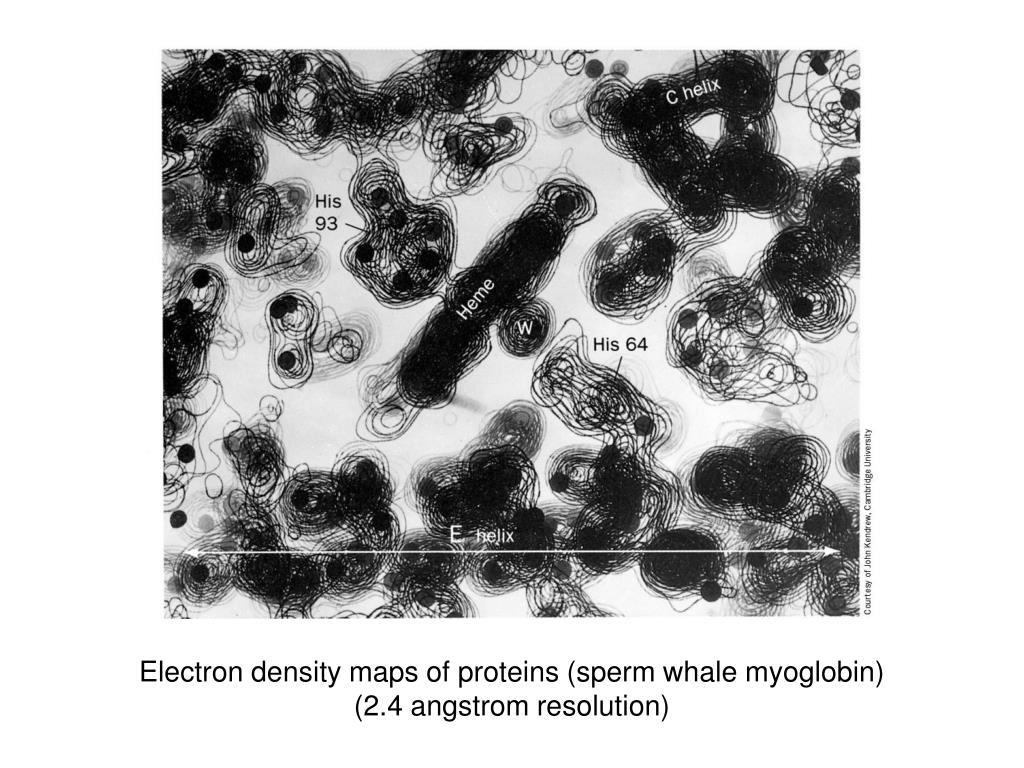
The terrain analogy is great - this is how the first structural biologists had to view electron density:
https://image.slideserve.com/105608/electron-density-maps-of...
I think this is a stack of 2d "slices" through the density, printed on transparencies. But I've also seen 2d representations of charge density (some of which is just theoretical studies) that are even more similar to a terrain map.
Neat. I've been exploring visual representations of electron density, optimized for educational use with XR. Intending to illustrate that science education content could be made transformatively less wretched.
Isosurfaces and nonlinear color gradients are educationally problematic. And fuzz gradients are hard to visually stereo fuse. And on near-term AR displays, black is transparent, and background environment colors can't be manipulated, which limits the space of attainable colorizations.
My current thought is to approach it as a volumetric light source. Composed of several linear scales, each a different color, with density as transparency. And respecting their true density when combined, so higher density scales "shine through" lower. We'll see. Maybe overlay random sample "sparkles" to maybe improve sense of spatial extent.
I've not seen much similar work. If you know of any, I'd love to hear of it. Also, it seems there was a push some years back to emphasize electron density when teaching intro chemistry. A push that doesn't seem to have gelled. I've guesses, but I'd value thoughts on why.
I've been out of the field for a few years, but I know that the graphics hardware (and accompanying software tools) make what you're describing much easier. This is best example I can come up with: https://jcheminf.biomedcentral.com/track/pdf/10.1186/1758-29... (see page 12)
Random asides:
1. once you're used to it, the simple isosurface mesh is super easy to work with (i.e. build molecules into), and for structural biologists the broad approximation is perfectly adequate
2. even so, the amount of unexplained blobs - some of which are definitely not noise - in my (xray) electron density maps was always both fascinating and frustrating. that's one reason why we don't spend much time on fancy renderings of experimental density at anything worse than subatomic resolution, because you're just sharpening the features that your atomic model doesn't explain.
> once you're used to it, the simple isosurface mesh is super easy to work with
One wouldn't know it from the state of content, or even of much education research, but educational representation is much harder than professional. Students are unable to untangle features reflecting careful correctness, from artistic license. So both seed mis/conceptions. Professionals can downregulate misconceptions... though that can be surprisingly localized - asking first-tier astronomy graduate students "What color is the Sun?"... gets you a common misconception.
Consider that ball-and-stick wrapped with electron density in Fig 9. Imagine coming at it cold. What is that stick? Well, maybe it's a ridge in electron density, thus symbolizing the surrounding not-so-stick-shaped region of increased density which constitutes a bond. But then what are those balls? They're way too similarly sized to be electron density. Ok, maybe they're spherical crosshairs for nucleus location, and the stick is a linear crosshair for the ridge. Variously sized because... something. Oh, no, some of the sticks are doubled, and density certainly doesn't have two ridges, or (here) increased density. So we've a paper notation that badly misrepresents actual electron distribution, blended into a physical representation with a... my head hurts.
I saw a professional chem ed content discussion yesterday, around a misconception, that in a two-species ionic solid, one atom bonds to another, in pairs. Rather than to "4 to 6" neighboring atoms... because one common printed diagram draws 4 neighbors, and another 6. It was like they were non-scientists, thinking they could wordsmith their way to correctness, without the slightest need to examine the actual characteristics of the real physical systems being described, or to consult with someone deeply familiar with them. The focus remained on models, decoupled from reality. I see a lot of that.
Chemistry education research describes chemistry education content using adjectives like "incoherent". Maybe XR can serve as an excuse to do better?
All I can offer in response is vague, hand-wavy speculation about human perceptive and cognitive limitations and how this impacts scientific representations. Coming from a biology background, I'm very used to artistic license and gross oversimplifications - and from years of habit, it almost hurts my brain to think about molecules as anything other than chicken-wire! From your complaint about those representations, I'm guessing you're unfamiliar with "Richardson ribbons": https://en.wikipedia.org/wiki/Ribbon_diagram
Even the developers of these visualizations would probably agree that it's dangerous to rely on them too much, and they are only a way to convey specific information in a way our eyes and brain can quickly process. Crystallographers in particular tended to over-rely on those chicken wire views and that plus software limitations yielded a lot of very poor-quality structures with poor atomic packing. The developer of those ribbon diagrams (Jane Richardson) has done a lot of other work to educate the field about how to visualize packing and other molecular properties and avoid screwing up the analysis. Over the long term, I think constant self-criticism makes up for the occasional sloppiness in scientific research.
My own focus is more on science education representations. And on how badly they describe the represented physical systems. Emphasizing that this seems as much a science community failure, as an education community fail.
Consider an illustration of the solar system in some introductory astronomy content. Let say, that not atypically, it misrepresents sizes, positions, orientations, lighting, and Sun color. Thus creating and reinforcing misconceptions known to be a problem in K-12, in undergrad, and even on into astronomy graduate school. In contrast, consider a line representing an Earth-Mars transfer orbit. It seems unlikely that students will say think there's a material long pole there, and worry about it hitting satellites and cities. Students and teachers are both clear on the aphysicality of the line, but not of the Sun color. Hmm, though using a minimum-energy line to represent an energy landscape is a source of misconceptions.
Similarly, common representation of atoms and molecules are known to be causatively associated with the stew of misconceptions that pervade students and teachers of chemistry. Ribbons, perhaps not so much. Hmm, though also similarly, they're often used without indication of regional flexibility, and so perhaps contribute to the underappreciation of configuration landscapes, of the importance of tuned floppiness. Perhaps.
Consider a currently implausible goal, of science education which accessibly describes the physical world, and conveys a transferable understanding of it. Arguably its content would look much more like scientific visualizations than content does at present. But being for education, it faces additional constraints, challenges and tradeoffs, distinct from those of professional scientific visualization. Creating such would require a collaboration of both deep scientific and educational expertise. For which very little incentive exists at present.
I'm trying to come up with an XR-compatible visual representation of electron density, that is physically correct, accessible, and bears in mind patterns of misconceptions in chemistry education. That task should probably be more than one random software dev's lockdown hobby hack. But as far as I know, that's were we're at. The related NSF-funded work I've seen... doing this bit well wasn't their focus. Same with XR ed tech side. Same with chem ed apps. And scientific visualization programs. And chem ed research. There may well be something nice out there, but I've not yet seen it. And big ed side... I was chatting with a leading textbook publisher, which onboards content creators with the indoctrination that their liberal arts background, and complete unfamiliarity with science and tech, is not a problem... because there's "a scientist" on call. Deriving electron density, with current nice python libraries, and GPUs, has surely gotten vastly easier than it was with old fortran messes, but still... it seems something more is needed here. Some societal staffing seems missing. No?
Mostly correct, except it's only indirectly the surface of the "atom" - because of the way covalent bonding works, there's continuous electron density between atoms too. It's just that the region around the nucleus is the most electron-dense. And yes, those look like phenylalanine or tyrosine (amino acid) sidechains to me, both of which have a benzene ring at the core.
(For the record, there is also something called neutron crystallography, which works like X-ray crystallography except it's the nuclei that diffract, not the electron clouds - which allows you to visualize hydrogen atoms more directly, and even resolve the difference between hydrogen and deuterium. But it's another speciality technique, in part because neutron sources are so much weaker.)
EDIT: since I actually did some work on visualizing electron density maps on the web, here's an interactive view of an older X-ray structure (from 2003, at only 3Å resolution) that shows how a protein molecule fits into the density:
http://natechols.github.io/xtal.js/map_viewer.html
(click and hold the middle mouse button to pan through the molecule)
Very good question. It'd be great to understand how this whole imaging process works. I have no idea, but it seems it involves many photos and rotation, and then DSP. So it's like CT for proteins .. maybe. It's like the x-ray crystallography but instead of repeating the x-ray many times on many-many samples. (edited)
Not many samples, many copies of the protein in what is often a single crystal (although if it's easily damaged, you might to use multiple crystals and combine the data post-processing). EM is actually very destructive, so they have to take thousands of micrographs of different individual molecules, and average them together.
Both techniques give you the average electron density over thousands to billions of molecules. The difference is in what kind of data you actually collect, and how you process and combine the data to get that averaged map:
- In X-ray crystallography, you're collecting the amplitudes of the Fourier transform of the electron density in the repeating unit of the crystal. The fact that it's a well-ordered (usually) crystal effectively "amplifies" the FT, and you get a series of images like this (from one orientation of the crystal):
https://cdn.britannica.com/02/147302-050-3F732246/X-ray-diff...
In either case, there's a lot of complex math that goes into reconstructing the electron density - but the end result, the electron density map, is very similar (aside from the absence of crystal packing interactions in the EM map).
I thought an individual atom has been imaged before, at least roughly. Anyway, this is fantastic science. I'm excited to see how imaging atoms affects our understanding of quantum mechanics and atomic structure in general.
I think they mean that this is the first time individual atoms have been imaged using this specific type of electron microscope. Individual atoms have of course been imaged as early as the 1980s using scanning tunneling microscopy. [1]
Last year, Aricescu’s team used cryo-EM to map the protein to 2.5 ångströms. But with the new kit, the researchers attained a 1.7-ångström resolution, with even better resolution in some key parts of the protein. “It was like peeling off a blur over your eyes,” Aricescu says. “At this resolution, every half ångström opens up a whole universe.”
You can, and it has been done. But that was with very small, flat molecules. Cryo EM can be used with very large molecules or complexes with many tens of thousands of atoms.
Feels like this might be useful for exploring the surface of cancer cells to identify receptors and other features that might be useful for developing targeted therapies.
At these scales, electrons behave more like waves than particles. Also the way that they are scattered by the subject and manipulated via em lenses causes information loss.
> On Oct. 11, 1955, Pennsylvania State University physics professor Erwin W. Müller and Kanwar Bahadur, who at the time was a Ph.D. student working with Müller, made history by being the first people to image individual atoms. The scientists were using a relatively simple and inexpensive instrument, and with it they directly observed individual tungsten atoms at the tip of a sharply pointed tungsten specimen.
I had heard once that new imaging modalities that advance a lot of new research invariably translate to Nobel prizes. Would that be expected of cryo EM techniques?
From the article: “That is a gold mine for structure-based drug design,” says Aricescu, because it shows how a drug could displace the water molecules, potentially resulting in medications with fewer side effects.
confirming certain molecular structures? We can nail down exact composition down to number of atoms in complex molecules, but the actual geometry is a (VERY well educated) "guesswork". edit1: well, its not actual guessing, there are other techniques that can also give hints for certain structural patterns (rings, chains, etc.) but still, its a very complex puzzle to piece the exact structure together.
We don't know yet. That is the idea about research.
When the microscope was invented, people made the same question: What is the use of it? Just watching things we already know bigger?
That is a recurring question. So much that Aristotle created the book MetaPhysics(meta meaning different from the Physics book) with the idea that this particular book did not need to be useful to be created. It was a compilation of non useful things.
This non useful compilation helped create science as we know it.
Oh please, this philosophical stuff is unnecessary. Cyro-EM is about a decade old and is just an alternative to X-Ray Crystallography for elucidating the structures of proteins. This article describes a way to improve the resolution of Cyro-EM, matching it to X-Ray Crystallography. The biggest bottleneck of X-Ray Crystallography is the production of protein crystals which is not necessary with Cyro-EM.
It's unbelievable how cryo-EM has come on. When i was a student, it was an also-ran behind crystallography and NMR, something that a few weird groups used to study a few special cases. Now you have to wonder if there's any point doing crystallography at all!
I still like NMR, because you're looking at proteins in solution. But if the freezing is good in cryo-EM, maybe even that becomes moot.
I'm not sure if it applies to proteins, but for smaller molecules there seemed to be some really interesting results a few years ago about using MOF's to adsorb analytes in a spatially repeating manner so x-ray diffraction techniques could be used without crystallization. Haven't followed closely enough to know if anything is panning out though.
It's especially shocking how many crystallographers have switched over - when I started, the ribosome crystal structures were the latest heroic achievement, but now nobody would waste time trying to crystallize ribosomes.
I agree about the microscope comments. On metaphysics I'm sorry but I have to disagree. "Metaphysics" was not Aristotle's name for those books, and unfortunately it's not even clear what the word means
https://plato.stanford.edu/entries/metaphysics/#WorMetConMet
There aren't any. The way cryo-EM works is it images hundreds of thousands of copies of the molecule and uses those to construct a 3D model, in this case, with atomic resolution. However, a single micrograph doesn't necessarily contain atomic resolution.
Haven't checked the article, but it's certainly possible the individual particle micrographs are archived somewhere. (But you're right, each will just look like a fuzzy gray blob.)
You can see that the record tests are being done with apoferitin or gaba. It all depends on the improved qualiry of direct detection cameras, filters, things like afis, fringe free imaging, ... The innovation goes insanely fast.
{kind=link}
{kind=link}
{kind=link}
Cryo EM is a very hot method right now to determine the three-dimensional structure of large molecules like e.g. proteins or protein complexes. Something like 10-15 years ago the best you could get was maybe 1.5 - 2 nm (these are rough numbers from memory), the results described in the article are 0.17 nm.